
🚀
A New Era for Medicines in Europe: The EU Pharma Package Deal Explained
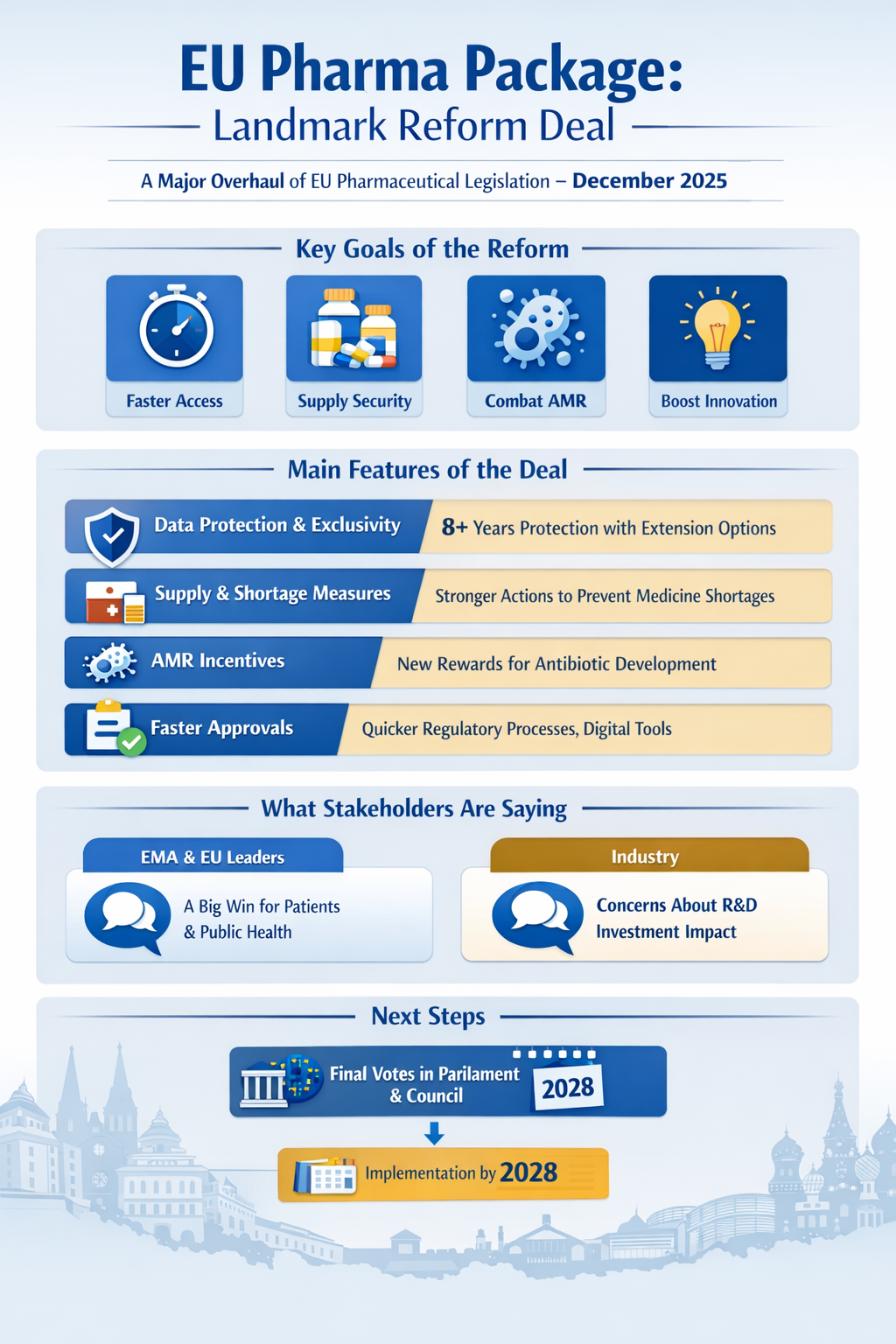
In early December 2025, after more than two years of intense negotiations, the European Commission, the European Parliament, and the Council of the European Union reached a historic political agreement on a comprehensive reform of the EU’s pharmaceutical legislation — commonly known as the “Pharma Package.”
This landmark deal is the most significant overhaul of EU pharmaceutical law in over two decades and represents a major milestone for patients, regulators, innovators, and public health systems across the Union.
Why This Matters
The EU’s current medicines framework has been largely unchanged since the early 2000s. Stakeholders have repeatedly called for modernisation to better address 21st-century public health priorities such as:
-
Faster access to new medicines and vaccines, especially innovative treatments
-
Better security of supply and fewer medicine shortages
-
Incentives for antibiotics and other priority therapies
-
Stronger regulatory cooperation and streamlining
-
Modern digital and scientific procedures
-
Tackling antimicrobial resistance and environmental impacts
All these themes are now anchored in the agreement.
What the Pharma Package Agreement Includes
1. Fairer Access & Stronger Innovation Support
The deal establishes updated data protection and market exclusivity rules designed to balance incentives for innovation with wider patient access:
-
Eight years of regulatory data protection — during which competitors cannot use original clinical data.
-
One additional year of market protection after authorisation.
-
Opportunity for further extensions for truly innovative medicines.
These protections help ensure innovators benefit from predictable market conditions while also fostering competition from generics and biosimilars once exclusivity ends.
2. Security of Supply & Shortage Prevention
To strengthen medicine availability across the EU, the agreement preserves provisions that empower Member States to:
-
Require companies to supply key medicines in sufficient quantities for patient needs
-
Introduce legal safeguards so that medicine shortages are reported early and managed proactively
These measures aim to curb the recurring problem of shortages that have plagued health systems in recent years.
3. Tackling Antimicrobial Resistance
The deal introduces new incentives for the development of priority antibiotics — including mechanisms like transferrable exclusivity vouchers that reward companies tackling antimicrobial resistance (AMR), a top public health priority.
4. Streamlined Regulation & Digital Transformation
The European Medicines Agency (EMA) and national regulators will operate under a reconfigured framework designed to be more agile and efficient:
-
Fewer scientific committees, with a simpler structure
-
Reduced assessment timelines (from 210 to 180 days)
-
Mandatory digital submissions and electronic product information formats
-
New regulatory “sandboxes” to test innovative approaches for cutting-edge therapies
These changes are intended to speed up authorisation processes while maintaining strict safety and efficacy standards.
What Stakeholders Are Saying
EMA & EU Institutions
The European Medicines Agency welcomed the agreement as a once-in-a-generation opportunity to modernise EU medicines regulation and better serve patients and public health goals.
EU Commissioners and legislators highlighted the deal as a boost to competitiveness, innovation, and resilience in the life sciences sector — signalling a long-term vision for high scientific standards and sustainable healthcare systems.
Industry Reaction
Not all reactions have been uniformly positive. Some industry voices warn that changes to exclusivity and regulatory conditions could impact incentives for R&D investment in Europe, possibly prompting companies to shift activities to markets with more favourable conditions.
What Happens Next
While the political agreement is a major milestone, the package still needs formal approval by both the European Parliament and the Council of the EU before it becomes law. Implementation will follow in stages, with many technical details and guidance documents to be developed — including delegated acts, implementing acts, and procedure manuals.
Most new provisions are expected to start applying from around mid-2028, after transitional arrangements and guidance roll-out.
Why This Reform Is a Turning Point
This Pharma Package represents more than just regulatory tinkering — it’s a strategic shift toward a more patient-centred, innovation-friendly and resilient pharmaceutical ecosystem in Europe. By rebalancing incentives, modernising procedures, and addressing systemic weaknesses like shortages and antimicrobial resistance, the EU aims to:
-
Bring safe, effective medicines to patients sooner, with less administrative friction
-
Ensure competitiveness of European pharma on the global stage
-
Build stronger public health systems capable of responding to future challenges
For patients, industry, and regulators alike, the deal signals a new chapter in Europe’s medicines policy that could shape health outcomes for decades.
At Regvista, we remain committed to support biopharmaceutical leaders navigating their transformative journey—bringing cutting-edge therapies to market faster and safer. Please feel free to contact us by submitting your enquiry to deployment@regvista.co.uk